Welcome to RetiGene !
RetiGene is a curated database of genes known to cause inherited retinal diseases (IRDs).
Using the navigation panel on the left, you can explore comprehensive information on genes, phenotypes, and a range of related analyses. The database is updated monthly with findings from newly published studies using PubMed and OMIM.
If you published a new gene-disease association or notice that a gene is missing or incorrectly annotated, please contact us directly at: mathieu.quinodoz[at]iob.ch.
The current version is v1.7 with curation of PubMed and OMIM until 25.11.2025.
You can download the data and figures here.


Gene curation has been led by Mathieu Quinodoz, Elifnaz Celik, Dhryata Kamdar, and Carlo Rivolta (IOB, Basel, Switzerland), in collaboration with ERDC-affiliated groups, including:
Manon Bouckaert, Marta Cortón, Emma Delanote, Lidia Fernández-Caballero, Gema García García, Lara K. Holtes, Marianthi Karali, Irma Lopez, Pilar Barberán-Martinez, Nina Schneider, Lieselot Vincke, Carmen Ayuso, Sandro Banfi, Beatrice Bocquet, Frauke Coppieters, Frans P. M. Cremers, Chris F. Inglehearn, Takeshi Iwata, Vasiliki Kalatzis, Robert K. Koenekoop, José M. Millán, Dror Sharon, and Carmel Toomes. Additional contributors from IOB include Francesca Cancellieri, Karolina Kaminska, Mukhtar Ullah, and Virginie G. Peter.
This website has been developed using the Shiny web application framework.
Genomic information (hg19)
Genomic information (hg38)
Figures:
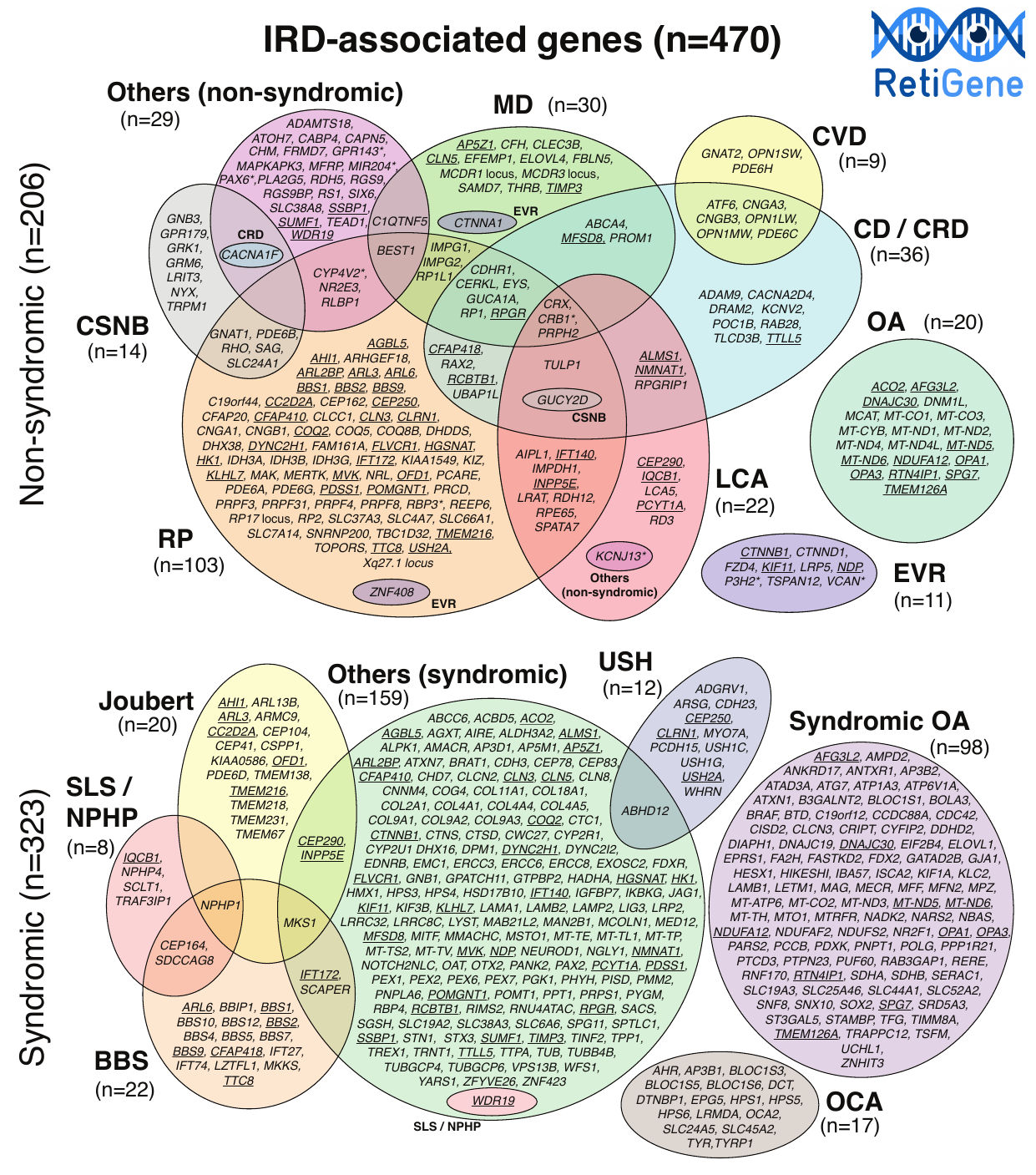
Genes and phenotypes
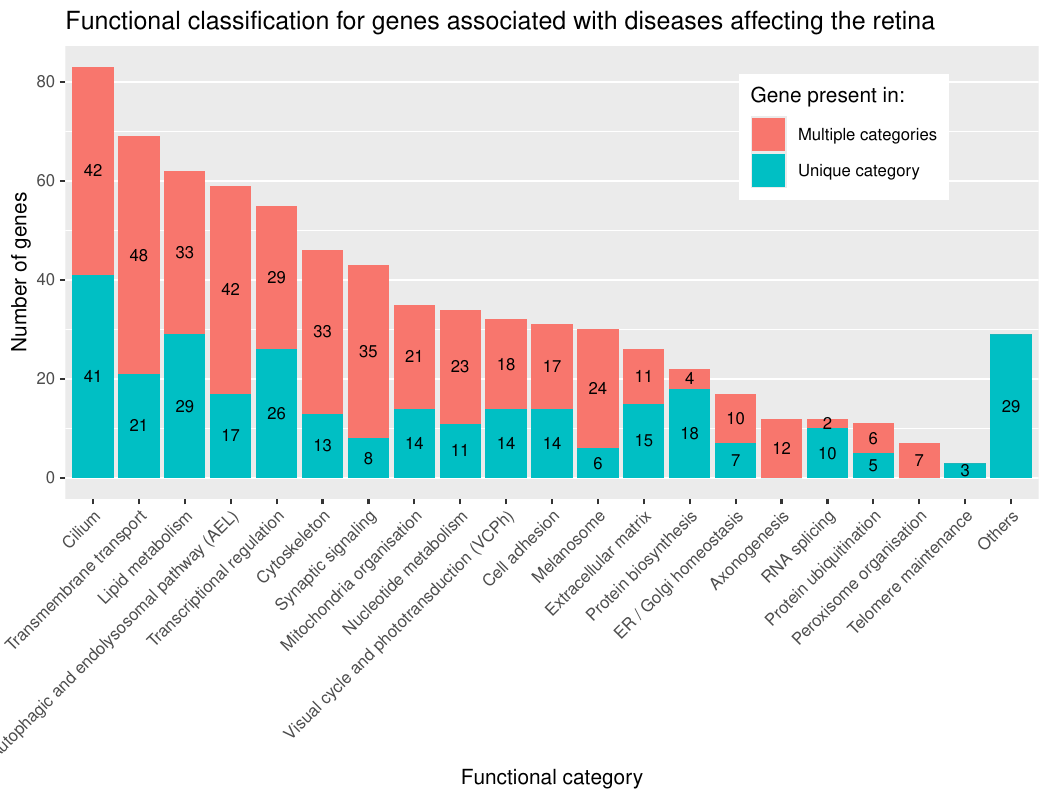
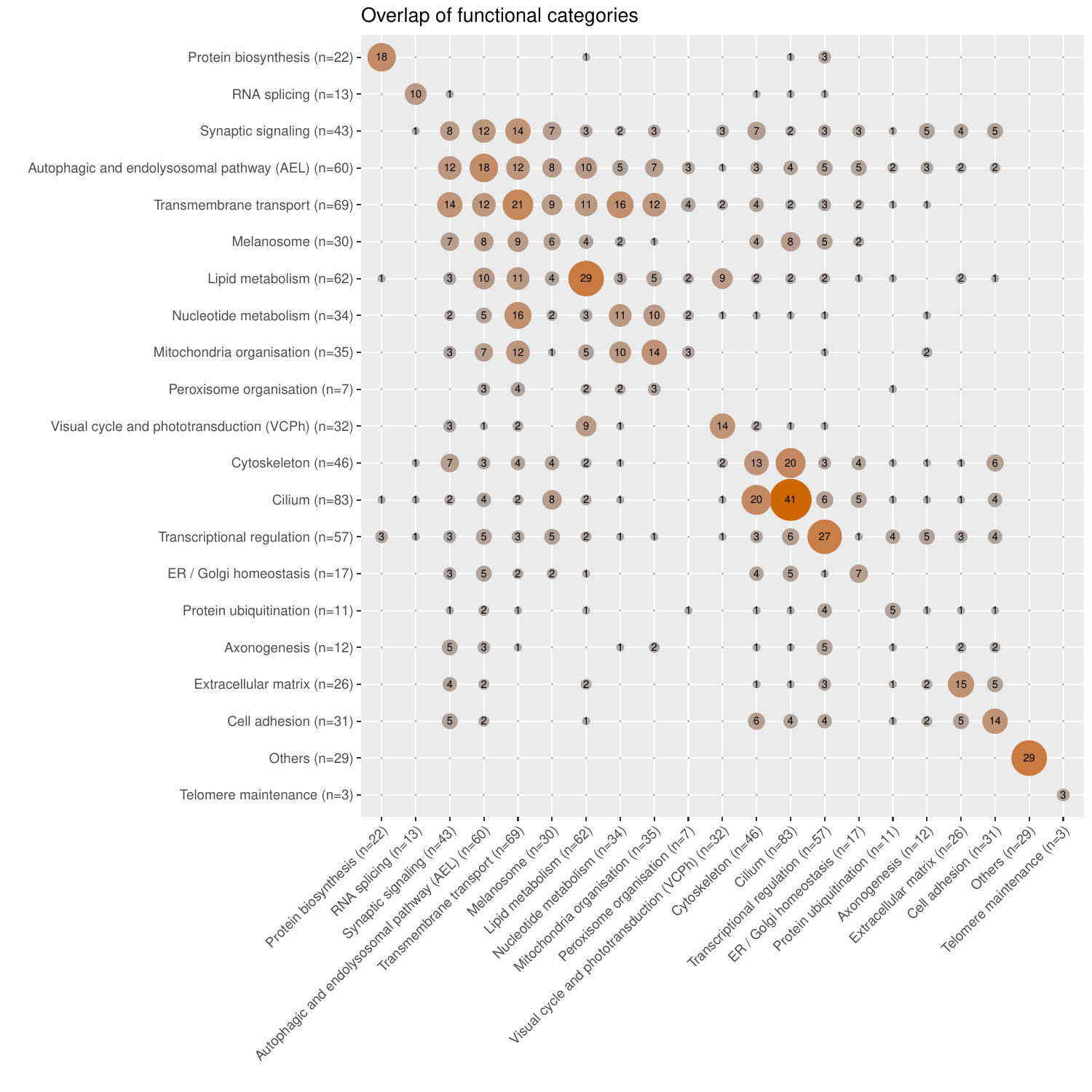
Functional annotation
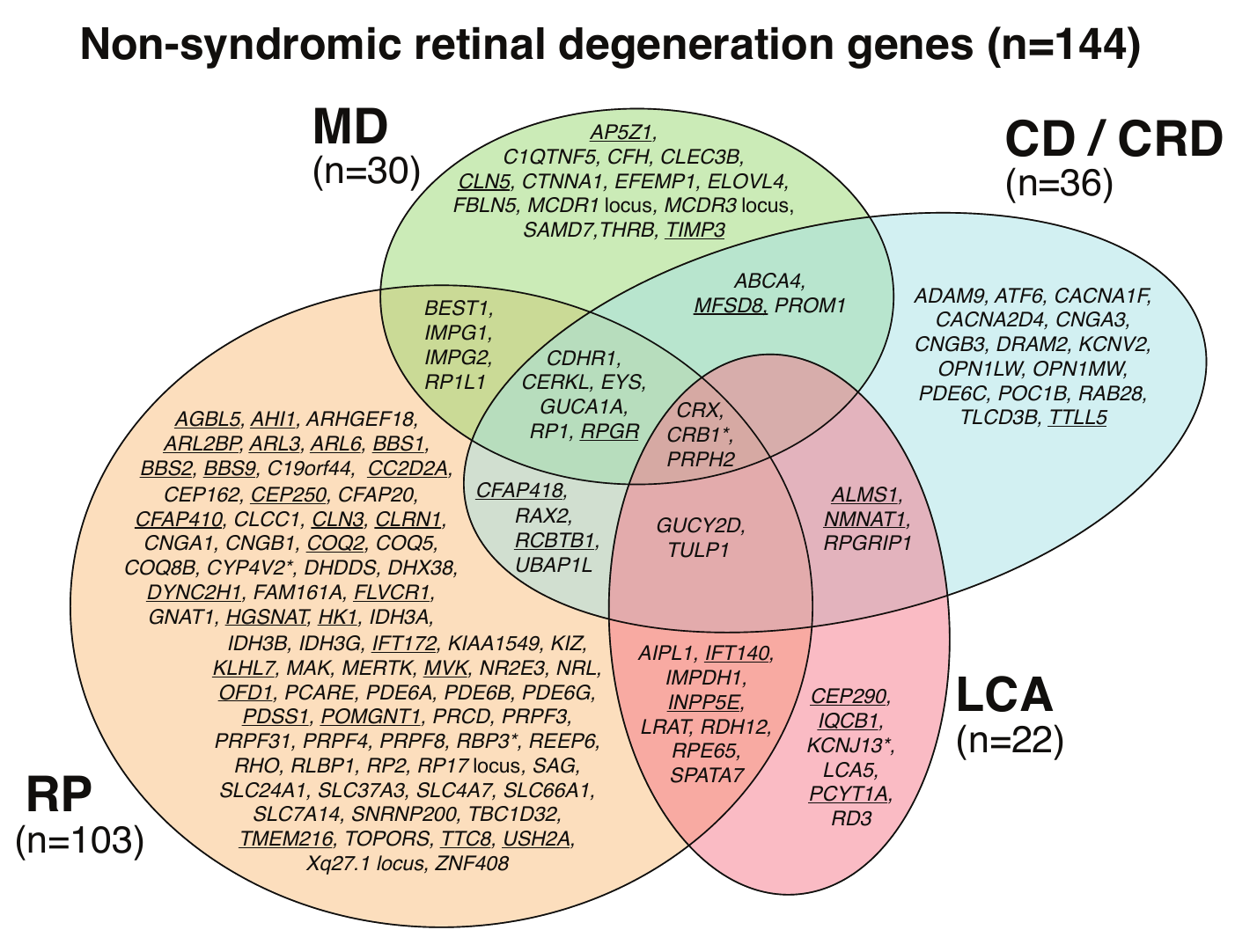
Non-syndromic retinal degeneration genes (most common genes)
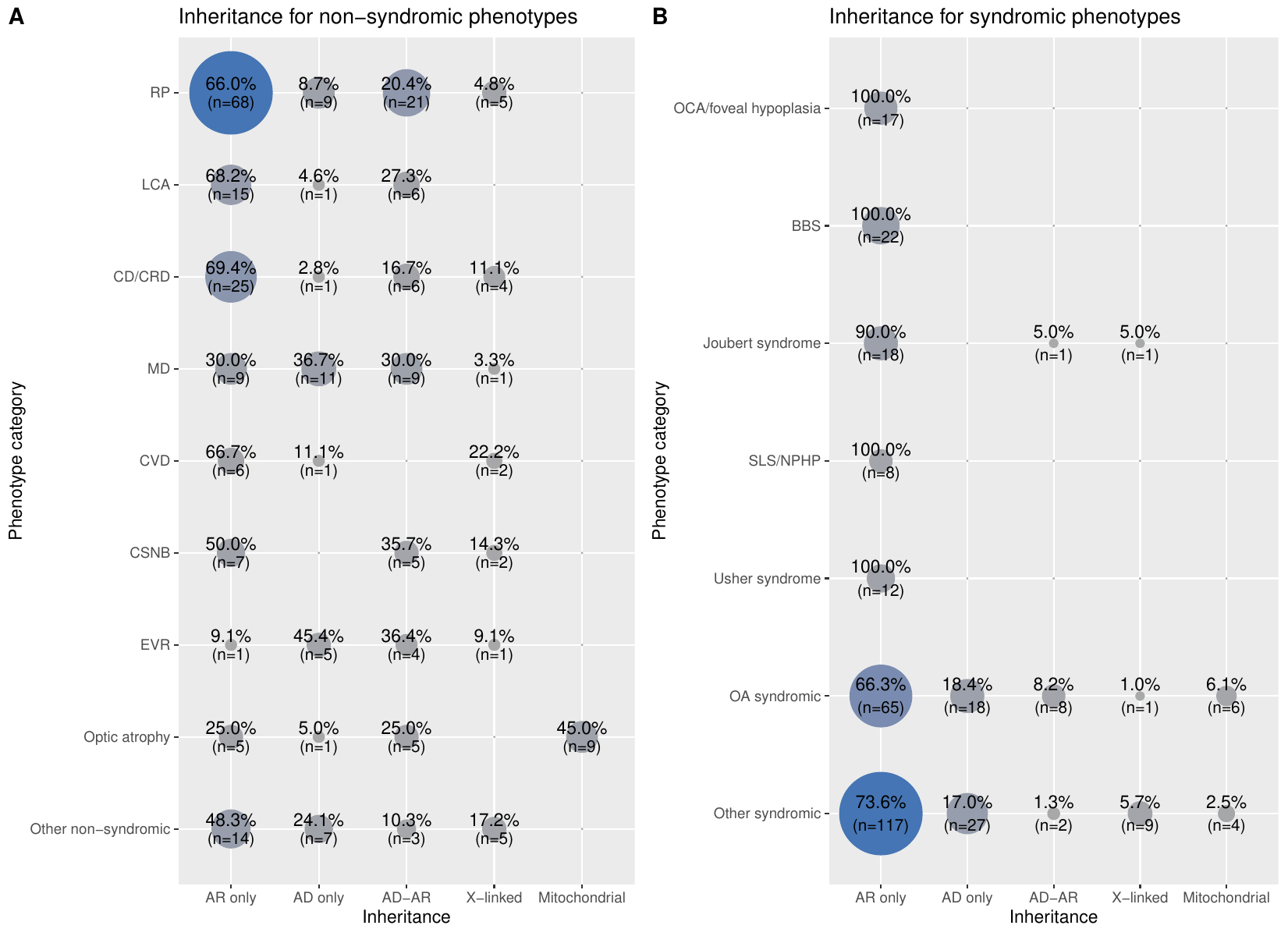
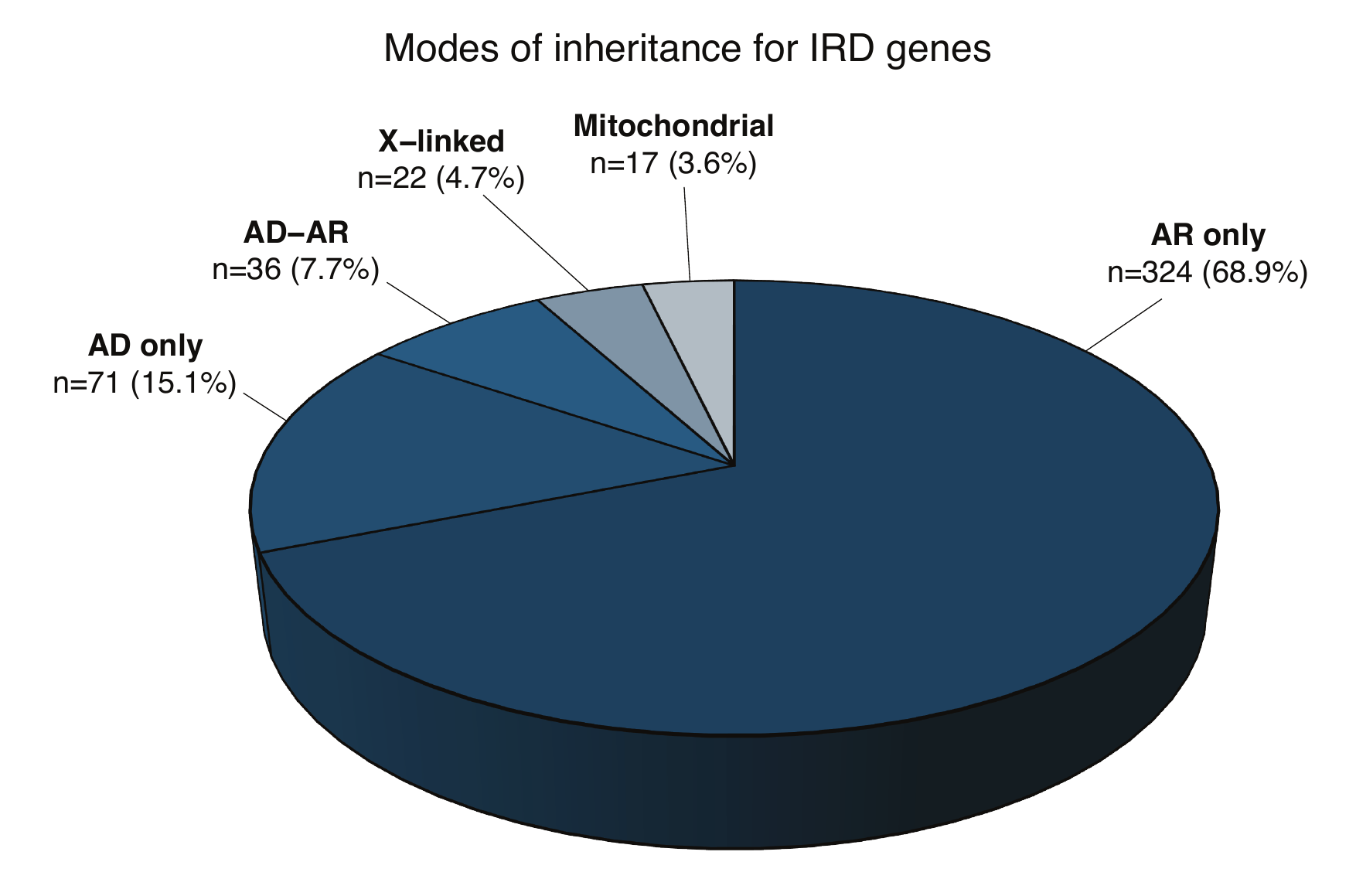
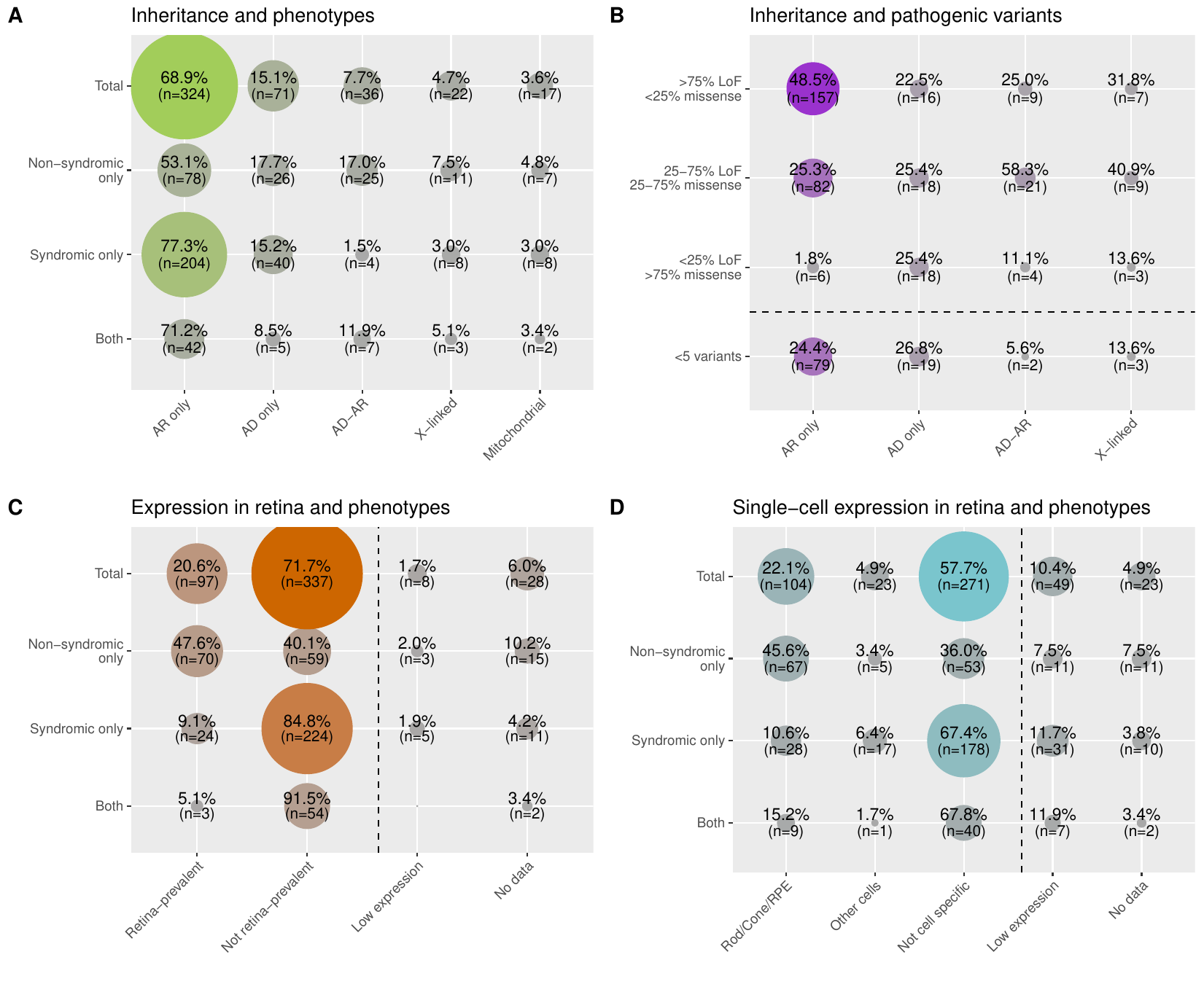
Inheritance for specific phenotypes
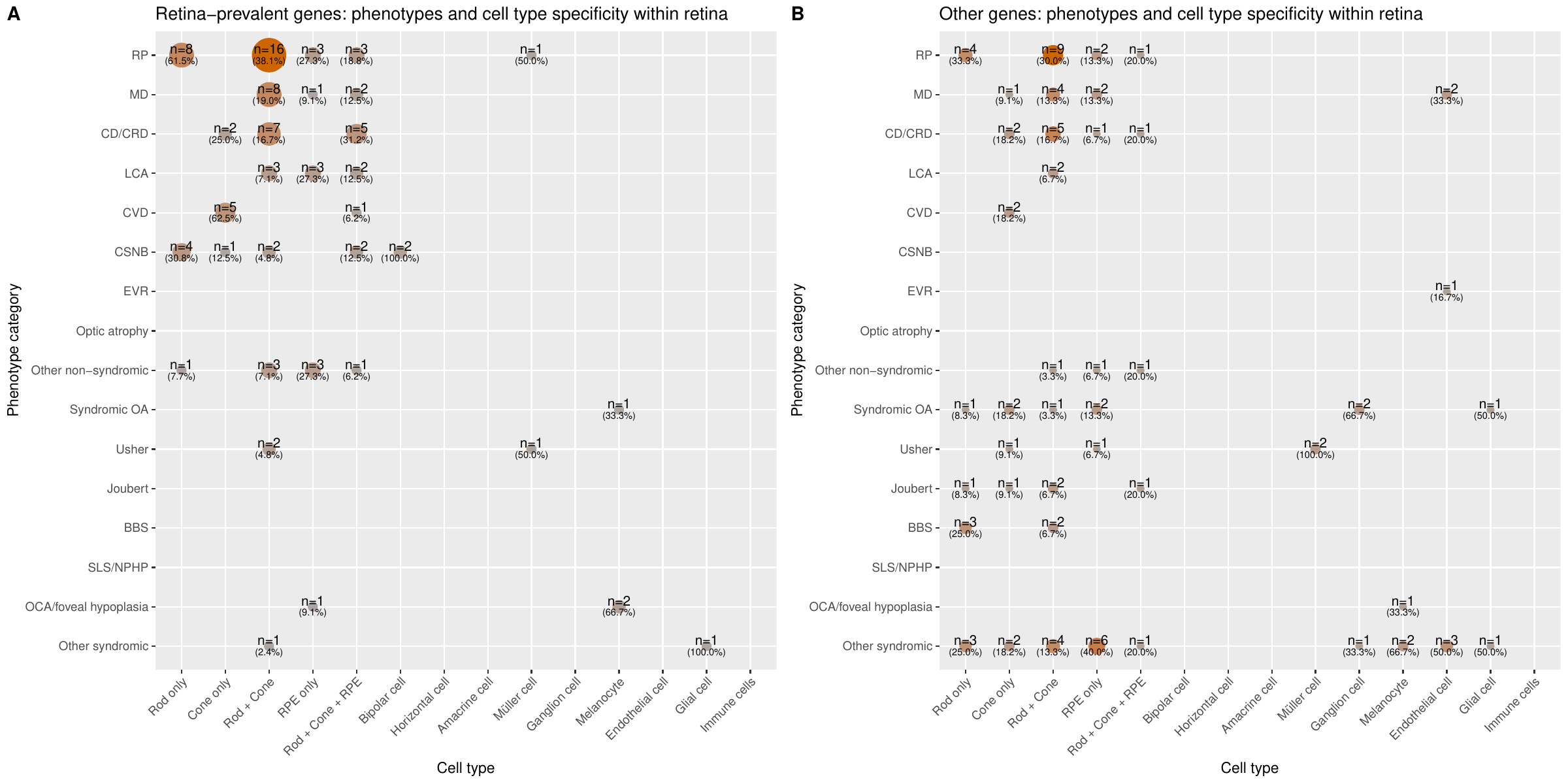
Cell-type specificity of expression
Candidate and unsupported genes
Supplemental Information
Genes
This table of genes is a catalog of all IRD associated genes, along with the year the gene was first linked to an IRD phenotype, its broad phenotype category, its inheritance mode, its sub phenotype category and its corresponding inheritance pattern, and the phenotype associated with it on the OMIM database. There are options to also look at the phenotype associated with the genes on OrphaNet (as seen on 3rd December 2024), the functional annotations, distribution of the pathogenic variants, and expression data of the gene at the tissue-level and at the scRNAseq-level within the retina.
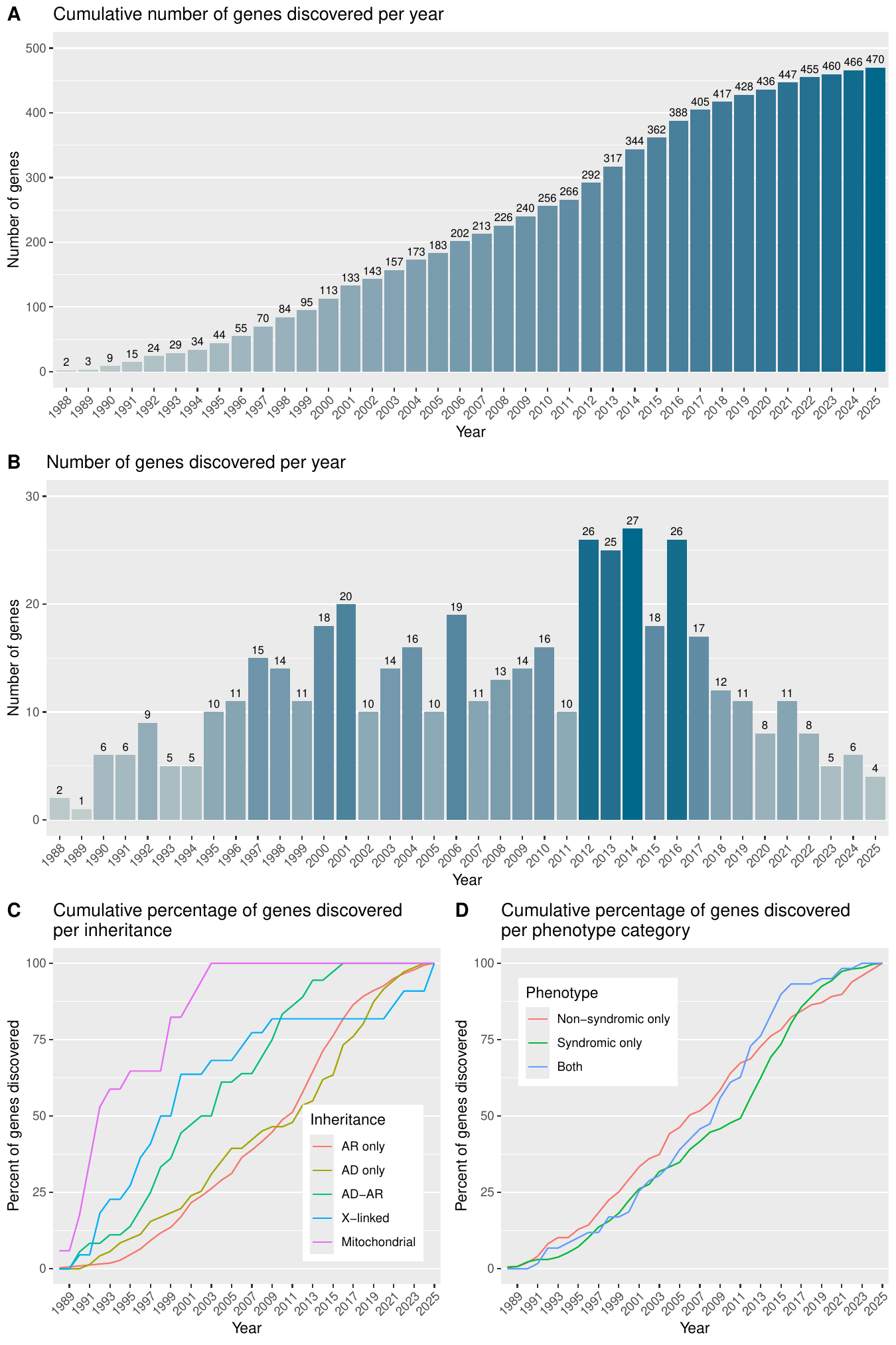
The initial list of genes associated with IRDs was compiled from multiple sources: OMIM, ClinGen (Retina GCEP, accession date 31st December 2024), LOVD (accession date 12th May 2023), Genomics England PanelApp (Version 7.0), RetNet, as well as reviews from the literature. This resulted in a preliminary list of 672 genes and loci. Each gene was independently assessed by two experts using the following criteria: (i) pathogenic variants observed in two or more families/unrelated individuals with consistent disease and inheritance pattern, (ii) same variant found in two or more families/unrelated individuals with solid functional data. Based on this review, 464 genes and loci met the inclusion criteria and were retained for downstream analysis. Another 192 genes were classified as “Candidates”, primarily due to evidence from only a single affected family. While 16 genes were “Discarded” due to insufficient evidence, conflicting data or definitive proof of non-association with IRDs. Loci identified from linkage and association studies without known causative variant(s) were excluded. If they had known causative variants, then they were checked for the criteria of inclusion as stated above.
- Gene name: gene symbol
- Year found: year of the first publication linking a gene with an IRD phenotype
- Broad phenotype category: states whether the gene causes a non-syndromic form of IRD (non-syndromic), a syndromic form of IRD (syndromic) or both (both).
- Inheritance mode: states whether the gene causes IRD in an autosomal dominant manner (AD), or autosomal recessive manner (AR), or both (autosomal dominant manner and autosomal recessive manner; AD-AR), or X-linked recessive/dominant manner (X-linked) or maternally inherited pattern (mitochondrial).
- Phenotype category (inheritance): 16 phenotype categories (derived from the Phenotype table) and their mode of inheritance for each phenotype.
- OMIM: phenotype according to OMIM database. It is not restricted to IRD phenotypes.
- Orphanet: phenotype according to Orphanet database with ORPHA codes. It is not resctricted to IRD phenotypes.
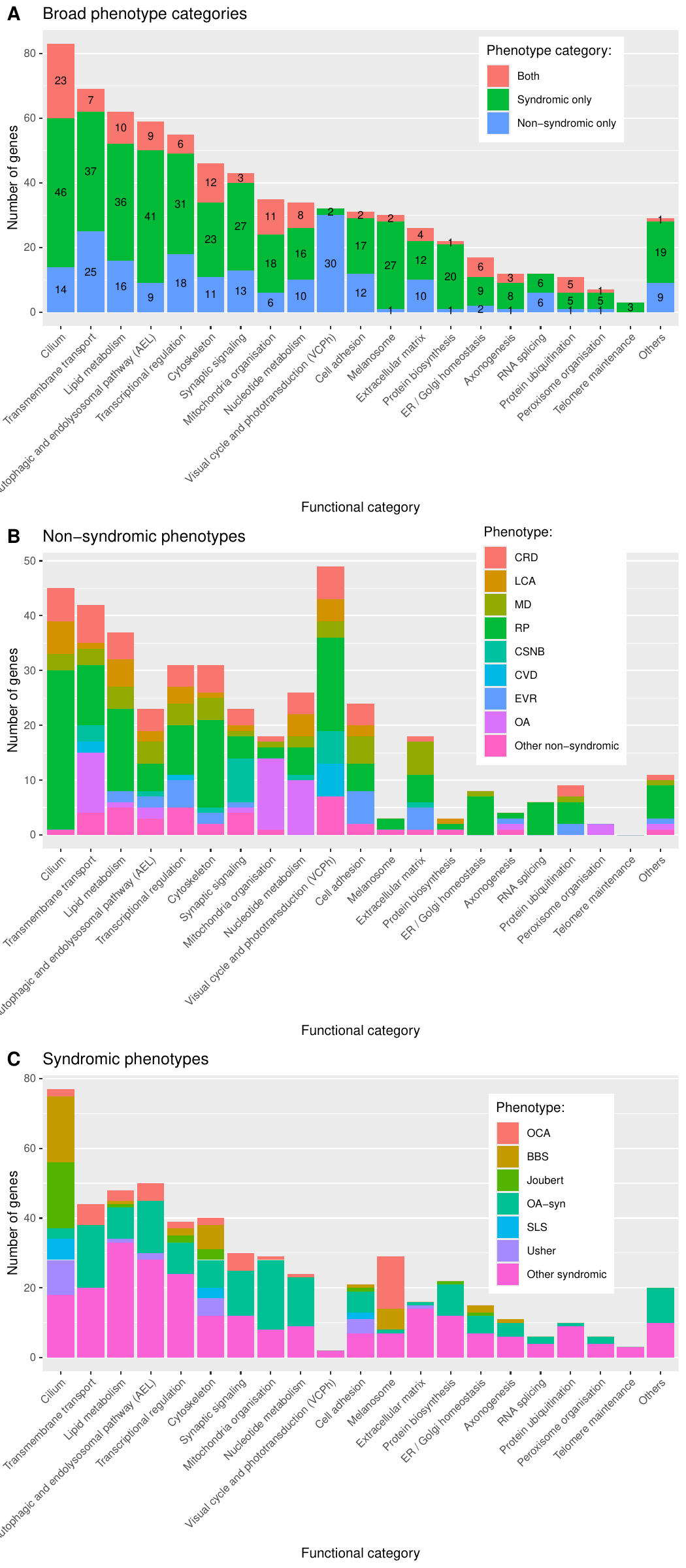
- Functional annotations: they denote which of the 23 categories (“Cilium”, “Lipid metabolism”, “Transmembrane transport”, “Transcription regulation”, “Cytoskeleton”, “Mitochondrial function”, “Visual cycle and phototransduction”, “Cell adhesion”, “Nucleotide metabolism”, “Endosome”, “Autophagy”, “Lysosome”, “Extracellular matrix”, “Melanocyte and melanin pathway”, “Ubiquitination”, “Spliceosome”, “Axonogenesis”, “Peroxisome”, “Protein biosynthesis”, “ER/Golgi homeostasis”, “Photoreceptor ribbon synapse”, and “Telomere maintenance” or “Others”), that we have intuitively chosen, the genes belong to. The categories are based on GO biological process terms (subset: GOTERM_BP_FAT categories; version of 2024-06-17 GO) along with manual classification through literature review. GO_FAT categories are only a subset of the more specific terms.
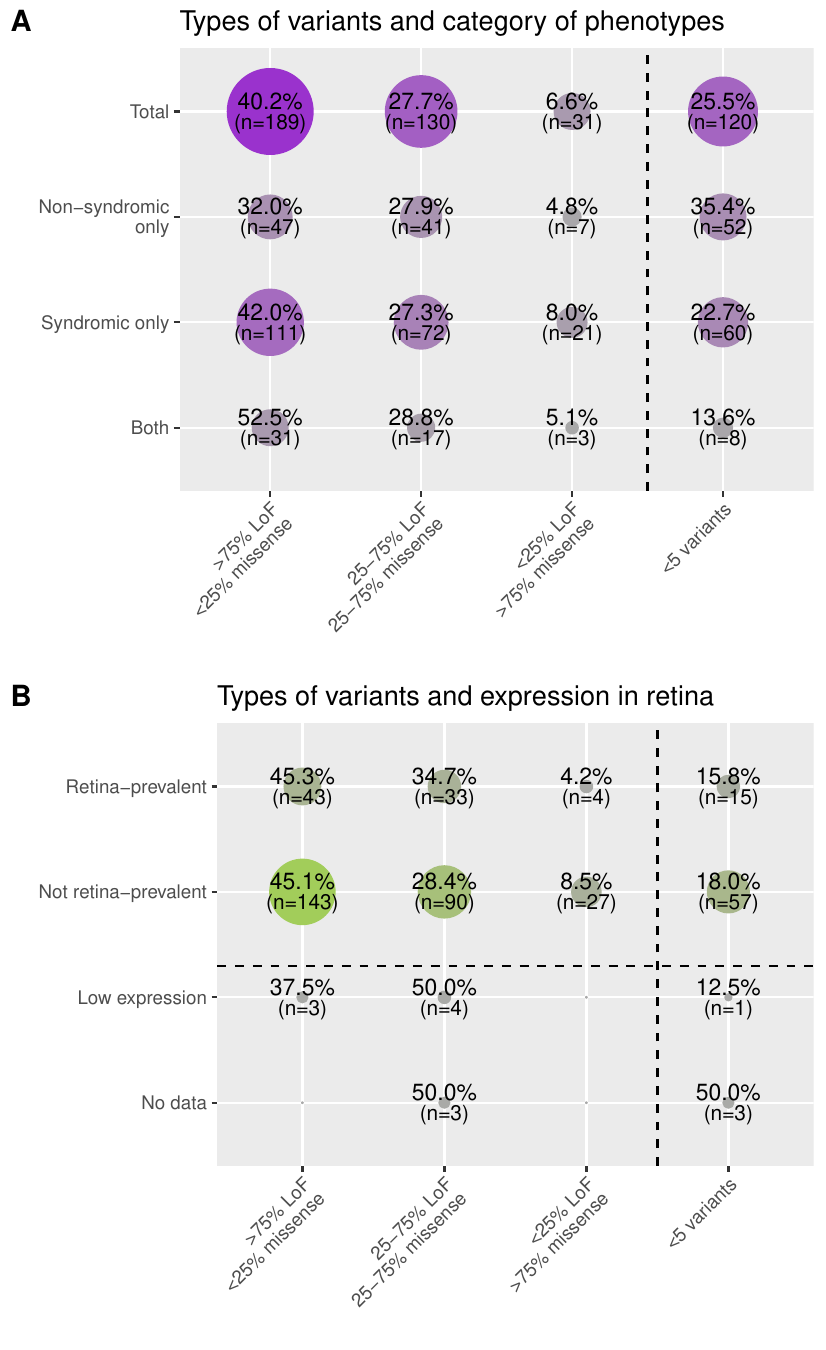
- Pathogenic variant types: the distribution of pathogenic and likely pathogenic (PLP) variants per gene from ClinVar (version of 21st January 2023) in four different categories: genes with (i) more than 75% of LoF PLP variants (>75% LoF <25% missense), (ii) mixed LoF and missense PLP variants (25-75% LoF 25-75% missense), (iii) more than 75% of missense PLP variants (<25% LoF >75% missense), and (iv) less than 5 PLP variants making them non interpretable (<5 variants).
- RNA-seq tissue: states for each gene whether it is highly expressed and specific in the retina compared to other tissues, based on FANTOM5 bulk RNA-seq Link. It is to be noted that skin and ear are not included in the FANTOM5 dataset. Z-scores and ratios from expression values are used to define 4 categories: “Retina-prevalent” means that the gene has high expression and specificity of expression in the retina compared to all the other tissues, “Not specific” means that the gene does not have high expression and specificity of expression in the retina compared to all the other tissues, “Low expression” means that the gene has low expression in the bulk RNA-seq dataset making interpretation inconclusive, and “No data’’ means the gene was not found in the dataset.
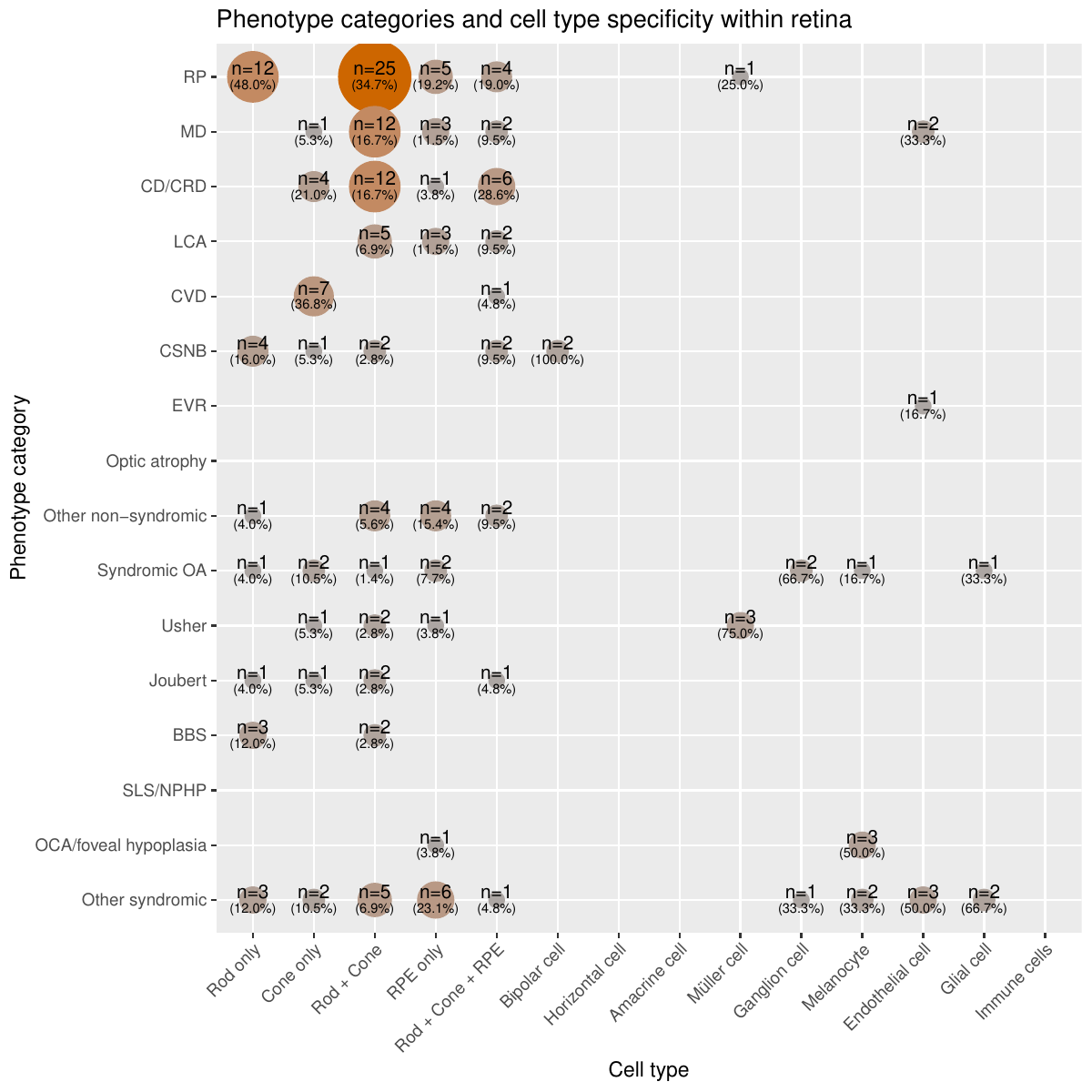
- scRNA-seq broad: states for each gene whether it is highly expressed and specific in the photoreceptors and RPE or other retinal cells in the retina. Retinal scRNA-seq data comes from Cowan et al.. Z-scores and ratios from expression values were used to define 3 categories. “Rod/Cone/RPE” if the gene passed the threshold to be highly expressed and specific to either rod, cones, or RPE or multiple of these three; “Other cells” if the gene passed the threshold to be highly expressed and specific to either horizontal cells, amacrine cells, bipolar cells, ganglion cells, Müller cells, melanocytes, endothelial cells, glial cells, or immune cells; “None” if it was found not to be highly expressed and specific to any of the retinal cell groups or if the gene has low expression in the scRNA-seq dataset making interpretation or if the gene was not found in the dataset.
- scRNA-seq cells: states for each gene whether it is highly expressed and specific to any of the 19 cell types or 5 larger cell groups in the retina. It is a more detailed version of ‘scRNA-seq broad’. If the gene passed the threshold for any of these cell types and groups based on the computed z-score and ratio of expression values, they were said to be specific and highly expressed in that cell type or group of cells. These 19 cell type groups are “Rod only”, “Cone only”, “RPE only”, “Horizontal cell”, “Amacrine cells”, “Bipolar cell”, “Ganglion cell”, “Müller cell”, “Astrocyte”, “Glial cell”, “Choroidal melanocyte”, “Microglial”, “Monocyte”, “NK cell”, “T cell”, “Mast cell”, “Pericyte”, “Fibroblast”, “Vascular endothelial cell” and the 5 larger groups are “Rod+Cone”, “Rod+Cone+RPE”, “Endothelial cells” (pericytes, fibroblasts, and vascular endothelial cells), “Immune cells” (NK cells, T cells, and mast cells), and “Glial cells” (astrocytes, glial cells and microglial cells). Additionally, they were marked “Not specific” if they were found not to be specific to any of the retinal cell groups, “Low expression” if the gene has low expression in the scRNA-seq dataset making interpretation inconclusive, and “No data” if the gene was not found in the dataset.
- References (PMIDs): PMIDs of relevant publications linking the gene to IRDs.
Genomic information (hg19)
A catalog of all the IRD associated genes, along with their broad phenotype category, their inheritance mode, their sub phenotype category and their corresponding inheritance pattern, and genomic position of the genes based on RefSeq. This allows one to look at genes within a desired genomic location.
- First 4 columns, see “Genes”
- Chromosome: chromosome in which the gene is present
- Start: beginning of the gene based on the longest RefSeq transcript (hg19)
- End: end of the gene based on the longest RefSeq transcript (hg19)
Genomic information (hg38)
Same as above but for hg38 genome build.
Phenotypes
Table listing the 16 phenotypic categories of IRDs and the number of genes associated to each category, along with the breakdown of what inheritance modes these genes follow.
- Phenotype: The 16 categories used to group the spectrum of diseases classified as inherited retinal diseases (IRDs).
- Total number of genes: number of genes linked to a particular phenotype.
- AR: how many of the genes linked to a particular phenotype have an autosomal recessive mode of inheritance.
- AD: how many of the genes linked to a particular phenotype have an autosomal dominant mode of inheritance.
- X-linked: how many of the genes linked to a particular phenotype have an X-linked mode of inheritance, be it dominant or recessive.
- Mitochondrial: how many of the genes linked to a particular phenotype have a maternal mode of inheritance. It is to be noted that genes can be both AR and AD, therefore the total number of genes per phenotypic category can be lower than the sum across the inheritance patterns.
Analysis
Candidate and unsupported genes
Table of genes that were not included in the list of genes associated with IRDs. This can as a result of insufficient evidence or because they were discarded due to novel evidence.
- Type: “Candidate” (insufficient evidence), “Candidate retinal phenotype in syndrome” (gene associated with a syndrome with enough evidence but too few cases display a consistent retinal phenotype), or “Discarded” (the gene-disease association was proven false based on novel findings such as population frequency from large databases for example.
- Reasons: details about why the gene was not classified as associated with IRDs.
- References (PMIDs): PMIDs of relevant publications.
List of acronyms used:
- BBS: Bardet-Biedl syndrome
- CD / CRD: Cone dystrophy, cone-rod dystrophy, Stargardt disease
- CSNB: Congenital stationary night blindness (with or without fundus albipunctatus)
- CVD: Achromatopsia, color vision abnormalities, color-blindness
- EVR: Exudative vitroretinopathy, Norrie disease
- LCA: Leber congenital amaurosis
- MD: Macular dystrophy
- OA: Optic atrophy, optic nerve hypoplasia
- OCA/foveal hypoplasia: Oculocutaneous albinism, foveal hypoplasia
- RP: Retinitis pigmentosa
- SLS / NPHP: Senior-Loken syndrome, nephronophtisis
- USH: Usher syndrome
- OA syndromic: Syndromic optic atrophy
- AR: Autosomal recessive
- AD: Autosomal dominant
- RPE: Retinal pigment epithelium
- RNA-seq: RNA sequencing
- scRNA-seq: single-cell RNA sequencing
- GO: Gene ontology
Downloads
You can download the data and figures here.
How to cite
You can cite the publication: Link.
Citation: Rivolta, C., Celik, E., Kamdar, D., Cancellieri, F., Kaminska, K., Ullah, M., Barberan-Martinez, P., Bouckaert, M., Cortón, M., Delanote, E., Fernández-Caballero, L., García García, G., Holtes, L.K., Karali, M., Lopez, I., Peter, V.G., Schneider, N., Vincke, L., Ayuso, C., Banfi, S., Bocquet, B., Coppieters, F., Cremers, F., Inglehearn, C., Iwata, T., Kalatzis, V., Koenekoop, R.K., Millán, J.M., Sharon, D., Toomes, C., and Quinodoz, M. (2025). RetiGene, a comprehensive gene atlas for inherited retinal diseases (IRDs), AJHG, 2025.
PMID: 40961941 DOI: 10.1016/j.ajhg.2025.08.017
Contact
Please send any comments, requests or suggestions to mathieu.quinodoz[at]iob.ch
Updates
10th June 2025 Version: 1.0
- Creation of the website
10th July 2025 Version: 1.1
- Update of the website with alternative gene names
- Possibility of downloading full gene table or only selected rows and columns
- Correction of gene name according to HGNC for GARIN4 (previously FAM71A)
- Correction of typo in now DNM1 (previously noted as DMN1)
- Addition of ATP1A3 (OA syndromic, AD) and FA2H (OA syndromic, AR)
- Addition of SNX31 as candidate gene (vitreoretinopathy, AD)
- Addition of OR2W3 as discarded
- Correction for MFRP (Other non-syndromic instead of other syndromic)
- Correction for CEP83 (Other syndromic instead of RP)
- Correction for ADAMTS18 (Other non-syndromic instead of CD/CRD)
- Update of PMIDs for MT-CYB, MT-TL1, and MT-TP
- Correction for IGFBP7 (Other syndromic instead of EVR)
- Correction for FRMD7 (Other non-syndromic instead of OCA)
- Addition of Non-syndromic OA for RTN4IP1
- Correction for SIX6 (Other non-syndromic instead of other syndromic)
- Correction for SSBP1 (Other syndromic and other non-syndromic instead of OA syndromic)
- Addition of RP-AR for BBS9 and related PMIDs
- Addition of genes: CREB3, NSUN3, BCAP31, RAB18, DOCK6, POLR3A, POLR3B, TMEM53, and RXYLT1
- New phenotype for TBC1D32, POC5, PCLO, and COQ8B
- New candidate genes: AARS1, RDH8, HSD17B4, IFT57, SOX5, NDUFV1, SMAD4, CD96, TBCE, and TBCK
- New candidate phenotype: PRPH2
- New discarded gene: LRP6
- Addition of genes: BCOR (RP, AR), and CRPPA (Other syndromic, AR)
- New candidate gene: BAIAP3 (RP, AR)
- New candidate phenotype: TOPORS (Other syndromic, AR)
- Addition of genes: ATRX (OA syndromic, X-linked), WDR73 (OA syndromic, AR), SURF1 (OA syndromic, AR), FKRP (Other syndromic, AR), LYRM7 (OA syndromic, AR), MINPP1 (OA syndromic, AR), SMPD1 (Other syndromic, AR), PPIB (Optic atrophy, AD), and LOXL3 (Other non-syndromic, AR)
- New candidate genes: NDE1, MTR, TGFBR2, COL6A6, CDK9, KATNIP, and SLC27A3
25th November 2025 Version: 1.7
- Addition of genes: HNRNPK (OA syndromic, AD), NDUFS7 (Optic atrophy, AR), NDUFV1 (Optic atrophy, AR), NDUFA1 (Syndromic OA, X-linked), NDUFA10 (OA syndromic, AR), NDUFAF3 (Syndromic OA, AR), NDUFAF4 (Optic atrophy, AR), and NDUFAF8 (Optic atrophy, AR)
- New candidate genes: NUP93, PCDH12, NDUFV2, and NDUFB11
- New phenotypes: CD/CRD (AR) for PCARE, and Joubert syndrome for RNU4ATAC
- Updated references: AIRE, and RP9